
 Généralités sur la Pharmacogénétique
Généralités sur la PharmacogénétiqueLa pharmacogénétique est une branche de la pharmacologie qui étudie l’influence du génotype sur la variabilité de la réponse à un médicament. En effet, certains individus recevant une dose standard de médicament ne vont pas présenter la réponse attendue au traitement, mais soit une diminution d'efficacité du traitement, soit des effets indésirables ou nocifs.
De nombreux analgésiques sont métabolisés par l'intermédiaire des iso-enzymes du cytochrome P450 (CYP) dont notamment le CYP2D6 soumis à un polymorphisme génétique. Les conséquences cliniques vont de la toxicité médicamenteuse à l'absence d'efficacité selon l'analgésique et le polymorphisme considérés. En détectant les polymorphismes génétiques par génotypage et/ou phénotypage, la pharmacogénétique permettra de mieux individualiser l'approche analgésique médicamenteuse et ainsi améliorer la sécurité et l'efficacité de nombreux antalgiques.
{kind=link}

Causes de variabilités:
Les variabilités entre les individus peuvent être dues à:
- Des états physiologiques particuliers
- Des co-morbidités
- Des facteurs environnementaux
- Des facteurs génétiques
Influence du génotype:
De nombreux polymorphismes génétiques peuvent affecter les gènes condant pour :
- Les transporteurs des médicaments
- Les enzymes métabolisant les médicaments
- Les enzymes ou récepteurs cibles des médicaments
Importance de la pharmacogénétique
Le but de la pharmacogénétique est de déterminer les profils génétiques des individus pour dépister ceux qui présentent un risque particulier de réponse inadéquate au traitement, à savoir d’inefficacité ou de toxicité vis-à-vis de certains médicaments. La pharmacogénétique permet d'individualiser la prescription de certains médicaments en pratique médicale courante afin d’optimiser les traitements, tant en termes d’efficacité que de sécurité d’emploi pour le patient.
Généralités en Pharmacocinétique:
On peut globalement distinguer 4 phases dans la pharmacocinétique d'un médicament:
1) son absorption
2) sa diffusion dans l'organisme
3) son métabolisme
4) son élimination de l'organisme
 Absorption d'un médicament
Absorption d'un médicamentLes différentes voies d'absorption:
voie orale ou per os
voie intra-veineuse* : sur une veine périphérique ou centrale
voie sub-linguale* : vers les veines linguales et maxillaires internes puis la veine jugulaire externe et la veine cave supérieure
voie rectale : vers les veines hémorroïdaires inférieures et moyennes puis en partie le tronc porte
voie sous-cutanée : généralement sur l'abdomen
voie cutanée ou trans-dermique*
voie intra-musculaire : quadrant supéro-externe du fessier ou deltoïde…
dans un organe ou in situ : intra-oculaire, intra-thécale, intra-tumoral…
voie nasale* (sprays) ou oculaire (collyres)
voie inhalée*
*Voies d'administration permettant d'éviter l'effet de premier passage hépatique
Modalités d'absorption/Biodisponibilité:
Le médicament doit passer une barrière qui le sépare de la circulation générale.
L'absorption est influencée par :
Les caractéristiques physico-chimiques du médicament, son pKa, son hydro/lipo solubilité, la taille et la morphologie de la molécule, la forme galénique (sirop, comprimé, gélule…).
Les caractéristiques liés à l'individu, son pH digestif, la vitesse de vidange gastrique et la mobilité intestinale, son alimentation, la prise associée de médicament, son âge, et ses pathologies associées (digestives, cardiaques...)
La biodisponibilité se définit comme la fraction de la dose de médicament administré qui atteint la circulation générale et la vitesse à laquelle elle l'atteint.
L'absorption digestive (la quantité de principe actif atteignant la circulation systémique) est difficile à mesurer puisque la circulation porte est d'accès peu aisé. L'approche de cette quantité se fait donc de manière indirecte à partir de la quantité de médicament dans le plasma prélevé au niveau périphérique, après le foie.
La quantité de médicament qui atteint la circulation générale est fonction de la quantité absorbée par l'épithélium digestif mais également, de processus d'élimination pré-systémique:
- dégradation dans la lumière intestinale
- métabolisme au niveau des entérocytes
- captage hépatique important au premier passage.
Si le médicament a une forte affinité pour l'hépatocyte et les enzymes hépatiques, une fraction de la dose absorbée est captée lors du premier passage avant d'atteindre la circulation systémique. La quantité de médicament retrouvée dans la circulation est alors diminuée. C'est l'effet de premier passage hépatique.
Distribution dans l'organisme
Une fois la circulation sanguine atteinte, les médicaments vont se distribuer dans l'organisme. Des caractéristiques physico-chimiques du médicament dépend son affinité pour les différents tissus, cependant d'autres facteurs vont influencer la distribution.
Dans la circulation générale, le médicament peut se lier aux protéines plasmatiques pour former des complexes. Il s'agit le plus souvent d'une liaison réversible et en équilibre selon la réaction suivante:
Médicament libre + Protéine libre ? Complexe médicament-protéine
Seul le médicament libre est actif!
Les différentes protéines plasmatiques et structures cellulaires impliquées dans le transport des médicments sont :
Albumine
Alpha1 glycoprotéine acide (AAG)
Lipoprotéines
Gammaglobulines
Cellules sanguines (érythrocytes, polynucléaires, lymphocytes, plaquettes)
La fixation aux protéines plasmatiques dépend beaucoup des caractéristiques acido-basiques du médicament.
Distribution dans l'organisme
Il existe un équilibre entre le plasma, les tissus et les voies d'élimination.
Lorsqu'un médicament fixé aux protéines plasmatiques est défixé, il est ensuite soit éliminé soit distribué vers les tissus. Si les voies de métabolisme sont efficaces, cette défixation n'aura le plus souvent aucune conséquence.
En pratique, la fixation protéique n'est à considérer que si elle est élevée (> 90 %) et si le médicament a une marge thérapeutique étroite (concentration toxique proche de concentration efficace).
Diffusion tissulaire:
Dans la plupart des cas, la distribution se fait dans l'espace extracellulaire, mais elle peut aussi comprendre le volume cellulaire. Pour diffuser, les médicaments doivent passer les membranes tissulaires. Dans certains tissus comme le foie, la paroi vasculaire est composée de capillaires discontinus ou fenestrés permettant une diffusion facile du médicament. En revanche, dans d'autres organes comme cerveau avec la barrière hémato-encéphalique, la paroi vasculaire est composée de capillaires continus difficilement franchissable.
Les mécanismes du passage trans-membranaire du médicament sont identiques à ceux de l'absorption digestive.
La diffusion tissulaire est donc dépendante de:
Caractéristiques physico-chimiques du médicament (lipophilie)
Capacité du médicament à franchir les parois vasculaires et cellulaires
La fixation protéique (sanguine et tissulaire)
Le débit sanguin tissulaire (très élevé pour le foie et le rein, faible pour l'os et la peau…)
Distribution dans l'organisme
Facteurs modifiant la distribution:
Volume liquidiens de l'organisme
Age (nourrisson…)
Déshydratation
Rapport masse maigre/tissu adipeux
Obésité
Age
Hémodynamique
Etat de choc
Insuffisance cardiaque chronique
Modifications des protéines plamatiques
Diminution de la concentration d'albumine
Grossesse
Syndrome néphrotique
Dénutrition
Grands brûlés
Cirrhose
Diminution AAG
Grossesse
Contraceptifs oraux
Age : nouveau-né
Cirrhose
Augmentation de la concentration AAG
Etats inflammatoires
Affections rhumatologiques
Etats infectieux sévères
Métabolisme des médicaments
Le métabolisme est la transformation, par une réaction enzymatique, d'une substance en un ou plusieurs autres composés actifs ou inactifs sur le plan biologique. De nombreux tissus peuvent réaliser cette transformation (peau, poumon, rein, intestin...), cependant le principal site de biotransformation est situé au niveau hépatique, dans les enzymes des microsomes. Ceci est en relation avec le flux sanguin très important du foie par rapport aux autres organes. Les hépatocytes contiennent un grand nombre d'enzymes impliquées dans la transformation des médicaments, en particulier les réactions d'oxydoréduction, les hydroxylations ou les ruptures oxydatives des liaisons N-C et O-C. L'élément fondamental de ce système enzymatique est le cytochrome P450 comprenant de nombreuses isoenzymes.
On peut distinguer 2 phases de métabolisme selon les processus de transformation induits par ces enzymes : [i]les réactions de phase I et celles de phase II.
Métabolisme des médicaments
Réactions de phase I:
Les réactions d'oxydation sont en grande partie localisées dans les microsomes hépatiques. Elles consomment du NADPH, de l'oxygène et passent par les cytochromes P450.
Les réactions de réduction sont beaucoup moins fréquentes. La réduction n'intervient pas exclusivement au niveau hépatique mais aussi dans l'intestin via la flore bactérienne.
L'hydrolyse est une voie métabolique banale, qui intervient dans différents tissus et même dans le plasma. Les enzymes du type des estérases sont le plus souvent non spécifiques. La réaction d'hydrolyse par clivage d'un ester ou d'un amide est très rapide chez l'homme,.
L'oxydation, la réduction et l'hydrolyse sont les biotransformations regroupées sous le terme de « métabolisme de phase I » et conduisent à des dérivés dont les groupements fonctionnels sont souvent des hydroxyles (-OH), des amines (-NH2) ou des carboxyles (-COOH).
Réactions de phase II:
Les groupements fonctionnels issus des réactions de phase I peuvent être ensuite conjugués. C'est la réaction de phase II.
Les mécanismes de conjugaison chez l'homme font généralement appel à l'acide glucuronique, à la glycine, au sulfate ou à l'acétyl.
Glucuroconjugaison. La conjugaison avec l'acide glucuronique est la conjugaison la plus fréquente. Elle est catalysée par la glucuronyltransférase et concerne les molécules possédant un groupement hydroxylé, carboxylé ou aminé. Les glucuronides sont très hydrosolubles ce qui explique leur bonne élimination dans l'urine et la bile. Dans certains cas, les esters sont instables et après hydrolyse dans l'urine ou le plasma redonnent la molécule mère.
Métabolisme des médicaments
POINTS IMPORTANTS CONCERNANT LE METABOLISME DES MEDICAMENTS
Lorsqu'un médicament est métabolisé, il l'est rarement de façon unique et plusieurs voies métaboliques sont possibles. Tous les métabolites ne sont d'ailleurs pas toujours identifiés
Il existe une spécificité pour les substrats : les différents cytochromes ont en fonction de leur structure protéique une affinité différente pour les divers substrats
Certains substrats modifient l'activité des enzymes responsables des biotransformations (inducteur ou inhibiteur)
Certaines enzymes des voies de métabolisme médicamenteux sont soumises à des polymorphismes génétiques qui peuvent modifier leur activité métabolique. On distingue alors des métaboliseurs lents, intermédiaires, rapides et même ultra-rapides.
Les médicaments qui ont une forte affinité pour les enzymes hépatiques ont, après administration orale, une faible biodisponibilité due à l'effet de premier passage hépatique.
Elimination
Elimination hépatique:
Le foie participe à l'excrétion des médicaments hors de l'organisme par le biais du système biliaire.
Après excrétion dans la bile, le médicament se retrouve dans la lumière intestinale où il peut être réabsorbé : c'est le cycle entéro-hépatique.
Elimination rénale:
La plupart des molécules sont éliminées dans les urines, soit inchangées, soit sous forme de produits de dégradation. Le plus souvent les médicaments ou leurs métabolites ont une masse moléculaire inférieure à 5kDa et sont donc filtrés par le glomérule. Seule la fraction non fixée est filtrée.
La réabsorption tubulaire intervient tout au long du néphron. Il s'agit souvent d'un processus passif qui est influencé par le degré d'ionisation du médicament : seule la fraction non ionisée au Ph urinaire est réabsorbée. On peut utiliser cette propriété dans certains surdosages pour accélérer l'élimination du médicament en alcalinisant les urines pour bloquer la réabsorption.
Certaines molécules subissent aussi une secretion active, entre autres les cations ou anions qui sont sécrétés dans la lumière du tubule par des systèmes de transport spécifiques consommant de l'énergie et à capacité saturable. On peut donc observer des phénomènes de compétition.
Autres voies d'excrétion:
Les autres voies (salivaires, pulmonaire…) sont habituellement négligeables par rapport aux voies rénale et hépatique.
On soulignera l'importance de la voie lactée, qui peut donner des risques d'intoxications du nourrisson lors de l'allaitement.
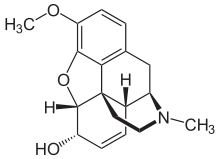
Codéine - Généralités
Propriétés Pharmacologiques:
MORPHINIQUE
ANTITUSSIF
ANALGESIQUE
SEDATIF
HISTAMINOLIBERATEUR
Mécanismes d'action:
Antitussif par inhibition du passage du stimulus tussigène au niveau des neurones de la medulla oblongata (centre de la toux), dans la partie dorsolatérale du bulbe.
Diminue les sécrétions trachéobronchiques.
Possède par ailleurs à dose élevée certaines autres propriétés de la morphine:
action analgésique, dépression du centre respiratoire, action spasmogène au niveau du tube digestif et des voies biliaires, augmentation du tonus des fibres musculaires lisses, action émétisante.
Effets Recherchés: (Par ls médecins petits galopiots, pas par nous autres junkies!
 )
)ANTITUSSIF
ANALGESIQUE
ANTIDIARRHEIQUE
Indications Thérapeutiques:
TOUX: Traitement symptomatique des toux non productives.
DOULEUR: Petit effet additif à celui du paracétamol
DOULEUR SPASMODIQUE
DIARRHEE: Traitement symptomatique.
Effets secondaires:
BOUFFEE VASOMOTRICE (CERTAIN RARE) Secondaire à l'effet histaminolibérateur.
HYPERSUDATION (CERTAIN RARE) Lié à l'effet histaminolibérateur.
ASTHENIE (CERTAIN RARE)
HYPOTENSION ARTERIELLE (CERTAIN TRES RARE)
HYPOTENSION ORTHOSTATIQUE (CERTAIN TRES RARE)
TACHYCARDIE (CERTAIN TRES RARE)
PALPITATION (CERTAIN TRES RARE)
ERUPTION CUTANEE (CERTAIN RARE) De type variable : exanthème, urticaire.
URTICAIRE (CERTAIN RARE) Le plus souvent dû à l'effet histaminolibérateur, plus rarement par hypersensibilité.
PRURIT (CERTAIN RARE)
ERYTHRODERMIE (CERTAIN TRES RARE)
ERYTHEME POLYMORPHE (CERTAIN TRES RARE)
ERYTHEME PIGMENTE FIXE (CERTAIN TRES RARE)
DYSURIE (CERTAIN TRES RARE) Par augmentation du tonus du sphincter vésical.
RETENTION D'URINE (CERTAIN TRES RARE) Par augmentation du tonus du sphincter vésical.
POLLAKIURIE (CERTAIN TRES RARE)
ANOREXIE (CERTAIN RARE)
NAUSEE (CERTAIN RARE) Par stimulation de la trigger zone du centre du vomissement dans l'area postrema.
VOMISSEMENT (CERTAIN RARE) Par stimulation de la trigger zone du centre du vomissement dans l'area postrema.
CONSTIPATION (CERTAIN FREQUENT) Par diminution du péristaltisme intestinal.
SPASME DU SPHINCTER D'ODDI (CERTAIN TRES RARE) Avec hyperpression des tractus biliaire et pancréatique, il peut s'accompagner d'une élévation des LDH.
TRANSAMINASES(AUGMENTATION) (CERTAIN TRES RARE) De mécanisme inconnu.
PANCREATITE AIGUE (A CONFIRMER )
SOMNOLENCE (CERTAIN RARE)
VERTIGE (CERTAIN RARE)
EXCITATION PSYCHOMOTRICE (CERTAIN TRES RARE)
EUPHORIE (CERTAIN RARE)
ANXIETE (CERTAIN TRES RARE)
CONFUSION MENTALE (CERTAIN TRES RARE)
MYOSIS (CERTAIN RARE)
MYOPIE (CERTAIN TRES RARE) Transitoire.
DEPRESSION RESPIRATOIRE (CERTAIN RARE) Par effet dépresseur sur les centres respiratoires du tronc cérébral. Elle n'entrainerait pas, aux doses analgésiques, de dépression respiratoire chez des sujets présentant une insuffisance respiratoire chronique obstructive (En revanche, aux doses "ludiques", cet effet est très présent...)
BRONCHOCONSTRICTION (CERTAIN TRES RARE) Secondaire à l'effet histaminolibérateur.
HYPOSECRETION BRONCHIQUE (CERTAIN RARE)
SECHERESSE DE LA BOUCHE (CERTAIN RARE)
REACTION D'HYPERSENSIBILITE (CERTAIN TRES RARE) A type d'éruption cutanée.
FRACTURE (A CONFIRMER )
APNEE (A CONFIRMER )
HYPERCORTICISME (A CONFIRMER )
Pharmaco-Cinétique:
- 1 - DEMI VIE 3 heure(s)
- 2 - ELIMINATION voie rénale
- 3 - ELIMINATION voie fécale
Absorption: Bonne résorption après administration orale.
Pic plasmatique en 1h environ après prise orale.
Répartition: Diffusion vers le SNC.
Passe la barrière placentaire.
Rapport salive/plasma voisin de 3 à 4.
Demi-Vie: 3 heures.
Métabolisme: Hépatique. Il existe 3 voies métaboliques: glucuroconjugaison, 0-déméthylation en morphine, N-déméthylation en norcodéine...
Élimination: Rein:
Élimination rapide, 2/3 en 6 heures, complète en 24h avec 40% de codéine libre ou conjuguée, 5 à 10% de morphine libre ou conjuguée, 10 à 20% de norcodéine libre ou conjuguée.
80% de la dose sont éliminés par voie urinaire, dont 60% sous forme de 6-glucuronocoide, 7% sous forme de morphine, 7% sous forme de norcodeine et 12% sous forme inchangée.
Fèces:
Traces.
Métabolisme de la codéine
La codéine est une prodrogue sans aucune activité analgésique. A peu près 10% de la dose de codéine absorbée est convertie dans l'organisme en morphine qui est un métabolite actif, par CYP2D6. La morphine est glucuronidatée en métabolites actifs et inactifs qui sont tous deux éliminés par les reins.
Le reste de la codéine est métabolisée par glucuronidation et CYP3AD4 en métabolites inactifs. C'est pourquoi CYP2D6 est indispensable à l'action analgésique de la codéine.
La plus grande partie des caucasiens convertissent rapidement la codéine en morphine via CYP2D6. Cependant, approximativement 7% à 10% des caucasiens possèdent un polymorphisme génétique responsable d'une perte d'activité de CYP2D6 et donc un métabolisme lent des substrats de CYP2D6 alors que 1 à 7 pour cent des caucasiens et plus de 25 pour cent des Éthiopiens ont une ou des duplications des gènes codants pour CYP2D6 et sont classés comme ayant un métabolisme ultra-rapide
Chez ces patients, la conversion de la codéine en morphine est réduite, comme son efficacité analgésique. L'administration de codéine à ces patients ne fournira pas le niveau attendu d'analgésie.
Les effets secondaires de la codéine sont également réduit lorsque l'activité intrinsèque du CYP2D6 est réduite. De même, les médicaments qui réduisent l'activité du CYP2D6 inhibent l'activité analgésique de la codeine.
En considérant que tous les inhibiteurs de CYP2D6 n'ont pas été étudiés en interaction avec la codéine, tous seraient susceptible d'interagir dans une certaine mesure.
Une grande partie de la dose de codéine absorbée est métabolisée par CYP3A4 et glucuronidation. Un patient
recevant de la codéine et un inhibiteur puissant du CYP3A4 pourrait s'attendre à métaboliser plus de codéine par l'intermédiaire du CYP2D6 et la voie de la glucuronidation. Ce processus pourrait entraîner une augmentation de la production
de métabolites actifs. Bien que les données soient limitées, les patients prenant un inhibiteur du CYP3A4 peuvent être plus sensibles à la toxicité à la codéine, en particulier si ils sont des métabolisateurs très rapide de la codéine en morphine.
CYP2D6 et opiacés
Les opiacés faibles comme la codéine, l'hydrocodone, l'oxycodone, ainsi que le tramadol, sont métabolisés, au moins en partie, par le CYP2D6, ce qui peut à la fois affecter leur efficacité et leur toxicité.
La codéine est un pro-médicament qui doit être activée en morphine par le CYP2D6 (O-déméthylation) avant d'atteindre son effet analgésique. Ainsi, elle est inefficace chez 6-7% des Caucasiens métaboliseurs lents (6, 7). Chez les sujets EM, la codéine était activée en morphine, alors qu'aucune production de morphine n'était détectable chez les PM (7). D'autres études ont étayé cette hypothèse (8, 9, 10). Le traitement de la douleur avec des médicaments contenant de la codéine va donc avoir des effets limités chez les patients PM, et leur demande de doses élevées de codéine sans réponse clinique peut être prise à tort pour une dépendance à ce médicament. A l'inverse les métaboliseurs ultrarapides pourront voir l'effet de la codéine s'accentuer, la production de morphine étant augmentée.
Par analogie, d'autres dérivés de la codéine pourront souffrir d'une variabilité dans l'efficacité analgésique comme par exemple l'hydrocodone ou l'oxycodone.
Il en va de même de l'efficacité opioïde du tramadol qui est structurellement proche de la codéine. Analgésique d'action centrale, il possède des propriétés pharmacologiques le différenciant toutefois des autres classes d'analgésiques. Le tramadol a une affinité faible pour le récepteur opiacé, par contre son métabolite M1, O-déméthylé via le CYP2D6, possède des propriétés analgésiques, et une affinité pour le récepteur opioïde m 200 fois supérieure à la substance mère (11). Les deux énantiomères du tramadol pourraient quant à eux exercer leur effet analgésique via l'activation de voies monoaminergiques inhibitrices centrales, en inhibant la recapture de sérotonine et de noradrénaline. En raison de ce double mode d'action, le tramadol est donc clairement différent des opiacés traditionnels, en terme de profil d'effets indésirables, et d'activité analgésique (12). L'activité du CYP2D6 a un rôle majeur dans l'effet analgésique opioide du tramadol, comme le confirment les études cliniques et expérimentales, bien que le tramadol, à la différence de la codéine, reste actif chez les individus PM en raison des propriétés monoaminergiques de la substance mère (13). Chez les métaboliseurs lents du CYP2D6 l'effet opioide s'atténue, alors que l'effet monoaminergique s'accroît. A l'inverse une augmentation du nombre d'allèles du CYP2D6 fonctionnel augmente la quantité de métabolite M1, confirmant le lien entre la production du métabolite et le génotype du CYP2D6 (14). Une étude prospective pratiquée sur 300 patients a évalué l'impact du génotype du CYP2D6 sur la réponse au tramadol en analgésie postopératoire après chirurgie abdominale. Les non-répondeurs étaient deux fois plus nombreux parmi les PM (46,7%) comparés aux EM (21,6%), et les PM ont eu 2 fois plus fréquemment besoin de réserves médicamenteuses en salle de réveil (43 vs 21%) et durant la patient controlled analgesia (PCA) (26,7% vs 11,6%). (15).
La méthadone est un opiacé fort qui peut être utilisée comme analgésique. Il existe des différences interindividuelles marquées dans les concentrations de méthadone après administration d'une dose unique (jusqu'à 17 fois) (16). Ceci s'explique en partie par l'activité des cytochromes, notamment le CYP2D6. Les concentrations au steady state ajustées au poids et à la dose de méthadone ont été étudiées chez 256 patients toxicomanes substitués par méthadone et génotypés pour le CYP2D6. Celles-ci ont été les plus élevées chez les PM, les plus basses chez les UM, et intermédiaires chez les EM. Par ailleurs 72% des PM ont reçu un traitement de substitution efficace (contre 40% des UM), et 50% des UM et seulement 28% des PM ont reçu des doses de méthadone supérieures à 100mg/j (17).
Les voies métaboliques de la biotransformation de la codéine.
La conversion de la codéine en norcodéine par le CYP3A4 et en codéine-6-glucuronide par glucuronidation représente habituellement 80 pour cent de la clairance de la codéine, et la conversion de la codéine en morphine par le CYP2D6 ne représente que 10 pour cent de la clairance de la codéine (flèches bleues). La morphine est ensuite métabolisé en morphine-6-glucuronide et en morphine-3-glucuronide. La morphine et la morphine-6-glucuronide ont des activités opioïdes (flèches vertes). Les glucuronides sont éliminés par le rein et sont donc sensibles à l'accumulation dans les cas d'insuffisance rénale aiguë. Ici est présenté l'exemple d'un patient (flèches rouges) ayant un métabolisme ultra-rapide de CYP2D6 en plus d'une 'nhibition du CYP3A4 à la suite d'un traitement avec de la clarithromycine et du voriconazole, et une accumulation de glucuronide en raison d'une insuffisance rénale aiguë. Les flèches rouges avec des lignes pointillées indiquent un faible niveau de lconversion ou d'élimination des drogues, les flèches vertes en pointillés indiquent un faible niveau de pénétration du cerveau, et les flèches épaisses indiquent des niveaux élevés.
Conclusion
La codéine est inefficace à des doses habituelles pour 7 à 10 pour cent de la population caucasienne en raison de l'homozygotie pour des allèles non fonctionnelles de CYP2D6.2. D'autre part, parmi les personnes qui ont le métabolisme ultrarapide, la prise de codéine peut entraîner une augmentation de la production de morphine.
Mis à part laduplication génique des allèles codant pour CYP2D6 , une interaction médicament-médicament pourrait contribuer aux effets toxiques observés certains patients. En plus de l'O-déméthylation en morphine, la codéine est N-déméthylé en norcodéine par CYP3A4 et subit également glucuronidation. Les patient traités concomitamment avec un macrolide et un dérivé azole, deux inhibiteurs connus du CYP3A4 peuvent donc avoir une clairance de la codéine réduite, ce qui augmenterait le risque d'un surdosage d'opiacés en association avec la duplication du gène CYP2D6.
Les effets opioïdes de la codéine sont liés à des concentrations plasmatiques de morphine produite après la prise de codéine. Dans le cas d'un métabolisateur ultra rapide ayant CYP3A4 inhibé, les concentrations sanguines des métabolites de la morphine, la morphine-3-glucuronide, et la morphine-6-glucuronide sont considérablement élevée. Le montant total de la morphine et de ses métabolites chez un tel patient correspond à 75 pour cent du montant total de la codéine présente dans son corps, tandis que le montant habituel de la morphine qui est produite après l'administration de doses multiples de la codéine atteint rarement 10 pour cent du montant total de la codéine chez une personne ayant un metabolisme normal de CYP2D6.
Les risques potentiels de dépression du système nerveux central après l'administration de petites doses de codéine chez un patient présentant un phénotype CYP2D6 ultra-rapide démontre l'utilité de la détermination du génotype et du phénotype dans la compréhension des réactions indésirables graves.
Le dextrométhorphane (DEM) est un dérivé morphinique non narcotique (composé synthétique analogue dextrorotatoire de la codéine) utilisé comme antitussif depuis une quarantaine d'années. Des données expérimentales indiquent que le DEM est dépourvu d'activité opiacée et qu'il agit in vivo comme antagoniste non compétitif du récepteur NMDA (N-méthyl-D-aspartate). Le DEM semble posséder à la fois une action neuromodulatrice et antinociceptive. Il est métabolisé en dextrorphane (DOR) par le CYP2D6, et il est utilisé comme substrat test (probe drug) pour phénotyper cette voie métabolique (18). Le DEM est rapidement transformée en DOR chez les EM. Les données expérimentales montrent que le DEM exerce un effet antinociceptif clair et marqué chez les PM du CYP2D6, au contraire de l'effet analgésique de courte durée observé chez les EM, et la courbe concentration plasmatique/effet chez les EM révèle une augmentation parallèle entre les taux plasmatiques de DEM et l'effet antinociceptif, alors qu'une relation inverse est observée pour le DOR (19).
Les voies métaboliques de la biotransformation de la codéine.
La conversion de la codéine en norcodéine par le CYP3A4 et en codéine-6-glucuronide par glucuronidation représente habituellement 80 pour cent de la clairance de la codéine, et la conversion de la codéine en morphine par le CYP2D6 ne représente que 10 pour cent de la clairance de la codéine (flèches bleues). La morphine est ensuite métabolisé en morphine-6-glucuronide et en morphine-3-glucuronide. La morphine et la morphine-6-glucuronide ont des activités opioïdes (flèches vertes). Les glucuronides sont éliminés par le rein et sont donc sensibles à l'accumulation dans les cas d'insuffisance rénale aiguë. Ici est présenté l'exemple d'un patient (flèches rouges) ayant un métabolisme ultra-rapide de CYP2D6 en plus d'une 'nhibition du CYP3A4 à la suite d'un traitement avec de la clarithromycine et du voriconazole, et une accumulation de glucuronide en raison d'une insuffisance rénale aiguë. Les flèches rouges avec des lignes pointillées indiquent un faible niveau de lconversion ou d'élimination des drogues, les flèches vertes en pointillés indiquent un faible niveau de pénétration du cerveau, et les flèches épaisses indiquent des niveaux élevés.
Conclusion
La codéine est inefficace à des doses habituelles pour 7 à 10 pour cent de la population caucasienne en raison de l'homozygotie pour des allèles non fonctionnelles de CYP2D6.2. D'autre part, parmi les personnes qui ont le métabolisme ultrarapide, la prise de codéine peut entraîner une augmentation de la production de morphine.
Mis à part laduplication génique des allèles codant pour CYP2D6 , une interaction médicament-médicament pourrait contribuer aux effets toxiques observés certains patients. En plus de l'O-déméthylation en morphine, la codéine est N-déméthylé en norcodéine par CYP3A4 et subit également glucuronidation. Les patient traités concomitamment avec un macrolide et un dérivé azole, deux inhibiteurs connus du CYP3A4 peuvent donc avoir une clairance de la codéine réduite, ce qui augmenterait le risque d'un surdosage d'opiacés en association avec la duplication du gène CYP2D6.
Les effets opioïdes de la codéine sont liés à des concentrations plasmatiques de morphine produite après la prise de codéine. Dans le cas d'un métabolisateur ultra rapide ayant CYP3A4 inhibé, les concentrations sanguines des métabolites de la morphine, la morphine-3-glucuronide, et la morphine-6-glucuronide sont considérablement élevée. Le montant total de la morphine et de ses métabolites chez un tel patient correspond à 75 pour cent du montant total de la codéine présente dans son corps, tandis que le montant habituel de la morphine qui est produite après l'administration de doses multiples de la codéine atteint rarement 10 pour cent du montant total de la codéine chez une personne ayant un metabolisme normal de CYP2D6.
Les risques potentiels de dépression du système nerveux central après l'administration de petites doses de codéine chez un patient présentant un phénotype CYP2D6 ultra-rapide démontre l'utilité de la détermination du génotype et du phénotype dans la compréhension des réactions indésirables graves.
CYP2D6 et antidépresseurs comme co-analgésiques
Les antidépresseurs peuvent être utilisés comme co-analgésiques, et ils ont montré leur efficacité en tant qu'analgésiques en situation aiguë ou chronique (20). Un certain nombre de mécanismes a été impliqué dans leur effet analgésique. Les antidépresseurs tricycliques sont connus pour inhiber le recaptage présynaptique de la noradrénaline (NA) et la sérotonine (5-HT) aussi bien que bloquer les récepteurs a1-adrénergiques, muscariniques et histaminiques. Les inhibiteurs sélectifs de la recapture de la sérotonine (ISRS) agissent principalement sur la 5-HT, et la venlafaxine, nouvel agent chimiquement distinct des tricycliques et des ISRS, inhibe la recapture de NA et 5-HT avec une action relativement sélective sur la 5-HT, particulièrement à faible dose. Les antidépresseurs sont associés à une incidence relativement élevée d'effets indésirables qui peuvent limiter leur utilisation chez les patients douloureux chroniques.
Plus de 25% des patients ne répondent pas aux antidépresseurs, et le phénotype UM est maintenant reconnu comme cause potentielle d'inefficacité thérapeutique entraînant chez certains patients la nécessité d'augmenter les doses pour être efficace (21). A l'inverse, d'autres expérimentent des effets toxiques aux doses usuellement recommandées. Ceci pourrait s'expliquer par la variabilité des concentrations plasmatiques (30 à 40 fois) observées suite à la prise de doses identiques (5).
Des études pharmacocinétiques indiquent que chez les PM, la clairance de nortriptyline, désipramine, imipramine, clomipramine, trimipramine et amitriptyline paroxétine, citalopram, fluvoxamine, fluoxétine et venlafaxine est diminuée et par conséquent leurs concentrations plasmatiques augmentées (4, 5). L'association entre les effets indésirables et le génotype du CYP2D6 est également bien étudiée. Une méta-analyse a montré une corrélation positive entre le risque de toxicité centrale (tremor, ataxie) et les concentrations plasmatiques médicamenteuses (22). Une fréquence augmentée d'allèles défectueux du CYP2D6 est retrouvée chez les patients présentant des effets indésirables (5). Dans une étude prospective, seuls les patients phénotypés PM pour le CYP2D6 traités par désipramine (100 mg/jour durant 3 semaines) ont présenté des effets indésirables (confusion, sédation, hypotension orthostatique) qui étaient par ailleurs corrélés à des taux plasmatiques élevés ayant nécessité une réduction de la dose (23). Une relation entre le phénotype PM et la cardiotoxicité (palpitation, souffle court, arythmie) a par ailleurs été suggérée chez les patients PM sous venlafaxine (5). Parmi les patients rapportant des effets indésirables sous antidépresseurs substrats du CYP2D6, on a retrouvé deux (24) à quatre (25) fois plus de PM. On note une variation des doses efficaces (dans le traitement de la dépression) de norptriptyline de 10 à 500 mg/jour, de 10 à 500 mg pour l'amitriptyline et de 25 à 300 mg pour la clomipramine selon que l'on soit PM ou UM (21). Les Chinois (majorité d'individus IM) métabolisent les antidépresseurs (désipramine, nortriptyline, clomipramine) plus lentement et les Orientaux reçoivent en général des doses plus faibles d'antidépresseurs (21). Ainsi, les premières recommandations de doses en fonction du génotype/phénotype du CYP2D6 chez les Caucasiens, basées sur des paramètres pharmacocinétiques et concernant 14 antidépresseurs, ont été développées en 2001 (26). La réduction de la dose est globalement de 50 à 80% pour les tricycliques chez les PM, et les différences sont moins marquées (30%) pour les ISRS. Pour les UM, les doses recommandées sont augmentées à 260% pour la désipramine, 300% pour la miansérine, et 230% pour la nortriptyline (26). Le génotypage pourrait en conséquence aider à individualiser les posologies et les intervalles entre les doses pour chaque patient.
Interactions médicamenteuses, phénotypage
et génotypage
et génotypage
Interactions médicamenteuses
L'activité des CYP peut également être modifiée par les interactions médicamenteuses.
Les substrats du CYP2D6 ne peuvent être induits, il existe par contre des inhibiteurs des substrats du CYP2D6, et lors de la co-administration d'un inhibiteur et d'un substrat du CYP2D6, on peut s'attendre à une augmentation des concentrations plasmatiques des antidépresseurs substrats du CYP2D6 ou encore à une inactivation des pro-médicaments comme les opiacés faibles (par la quinidine, les neuroleptiques comme la chlorpromazine, l'halopéridol, la lévopromazine, la thioridazine ou les antidépresseurs).
Phénotypage et génotypage
Certains tests sont dorénavant disponibles (phénotypage et génotypage), et utilisables en pratique clinique afin d'identifier les différentes activités enzymatiques ou les variantes alléliques. Ils sont une aide à l'individualisation de la thérapeutique afin de prédire la posologie adéquate du médicament approprié à chaque patient, en anticipant les effets indésirables ou l'inefficacité thérapeutique. Ils permettent par ailleurs de faire la distinction entre un problème de compliance et un métabolisme ultra-rapide, ou entre un abus médicamenteux (opiacés) et un déficit métabolique.
Le phénotypage est une mesure de l'activité enzymatique spécifique suite à l'ingestion d'un substrat test (probe drug) en mesurant son rapport métabolique (metabolic ratio MR) dans les urines (1, 27). Le phénotypage peut par ailleurs permettre de détecter des interactions médicamenteuses chez des patients dont le phénotype est connu au préalable, ou en réalisant en parallèle un génotypage.
Le génotypage permet l'analyse des mutations génétiques fonctionnelles importantes codant pour des enzymes spécifiques. Il est réalisé par polymerase chain reaction (PCR) couplée à une analyse par restriction fragment length polymorphism (RFLP) après hydrolyse avec l'aide d'enzymes de restriction (1). Il a l'avantage de permettre une détermination directe des informations génétiques de l'individu (réalisé une fois pour toute la vie) et n'est pas influencé par la prise concomitante de médicaments ou par les facteurs environnementaux. Le coût et la sensibilité du génotypage sont ses limitations actuelles. Il existe en effet des génotypes spécifiques ou des mutations encore inconnues, notamment pour le CYP2D6. Toutefois, les avancées sont rapides dans ce domaine et des méthodes de détection rapide par la technologie des puces génétiques sont actuellement commercialisées (CYP2D6 et CYP2C19) qui permettront d'analyser les variantes alléliques à plus grande échelle.
Cas clinique
Patiente de 55 ans connue pour un état dépressif majeur traité de longue date par paroxétine (Deroxat®) à raison de 20 mg/jour. La patiente présente des douleurs lombaires suite à chute, traitées par tramadol (Tramal®) à raison de 3 x 75mg/jour, inefficace selon la patiente. Malgré l'augmentation progressive des doses, la patiente ne note aucune amélioration et signale par ailleurs une impression ébrieuse, une confusion, une difficulté à coordonner ses mouvements, des tremblements et des sudations. Un génotypage du CYP2D6 est réalisé et compatible avec un bon métabolisme. Le phénotypage rapporte quant à lui un phénotype de métaboliseur lent.


Commentaire: Il existe en effet une interaction pharmacocinétique entre le tramadol et la paroxétine, qui est un inhibiteur puissant du CYP2D6, transformant le phénotype de la patiente de bon métaboliseur en métaboliseur lent, et donc à l'inefficacité opioïde du tramadol. Une accumulation de la substance mère du tramadol est attendue exposant ainsi au risque de syndrome sérotoninergique lors de l'association avec la paroxétine.
Conclusion et perspectives
Bonus:
 2000-2010 La codéine post-chirurgie met les enfants à risque Les enfants ont souvent prescrits de la codéine pour soulager la douleur après la chirurgie pour enlever leurs amygdales ou des végétations adénoïdes pour traiter l'amygdalite chronique ou d'apnée du sommeil, une condition dans laquelle les problèmes respiratoires font qu'il est difficile pour eux de dormir à poings fermés.
2000-2010 La codéine post-chirurgie met les enfants à risque Les enfants ont souvent prescrits de la codéine pour soulager la douleur après la chirurgie pour enlever leurs amygdales ou des végétations adénoïdes pour traiter l'amygdalite chronique ou d'apnée du sommeil, une condition dans laquelle les problèmes respiratoires font qu'il est difficile pour eux de dormir à poings fermés.
Cependant, certains enfants sont morts après avoir reçu la codéine en quantités qui sont dans la fourchette des posologies recommandées.
La Food and Drug Administration (FDA) prend des mesures pour mettre en garde contre l'utilisation de la codéine pour soulager la douleur des enfants après la chirurgie pour enlever leurs amygdales ou des végétations adénoïdes.
En Août 2012, la FDA a averti le public que ce danger existe pour les enfants qui sont des «métaboliseurs ultra-rapides" de la codéine, ce qui signifie que leur foie transforme la codéine en morphine en plus élevée que la normale.
Depuis lors, la FDA a procédé à un examen complet de la sécurité de la codéine chez les enfants. A la recherche de la FDA des événements système de rapports défavorables (SADR) base de données de 1969 à 1 mai 2012 a identifié 10 morts et trois surdoses liés à la codéine. Beaucoup de ces enfants ont été remis d'une intervention chirurgicale pour enlever leurs amygdales ou des végétations adénoïdes.
Fort d'alerte sera une nouvelle boîte d'alerte de la FDA être ajoutées sur l'étiquette des médicaments contenant de la codéine des produits sur le risque de la codéine pour soulager la douleur chez les enfants après une amygdalectomie et / ou adénoïdectomie. (L'étiquette d'un médicament est la documentation accompagnant un médicament d'ordonnance.)
La FDA recommande vivement contre l'utilisation de la codéine pour soulager la douleur chez les enfants après une amygdalectomie et / ou adénoïdectomie. L'agence demande aux professionnels de la santé d'utiliser un autre analgésique.
En outre, les parents et les soignants doivent être conscients des risques d'un traitement codéine après une amygdalectomie ou une adénoïdectomie et doit demander un médicament contre la douleur différente si leur enfant est prescrit codéine dans ce cadre
Le problème
La codéine est un analgésique opioïde analgésique narcotique des médicaments-une-utilisé pour traiter les douleurs légères à modérées. Il est également utilisé afin de réduire la toux, le plus souvent en combinaison avec d'autres médicaments. Codéine est disponible sur ordonnance, seul ou en combinaison avec l'acétaminophène ou de l'aspirine, et dans certains médicaments contre le rhume et la toux.
La codéine est convertie en morphine dans le foie par une enzyme. Certaines personnes ont des variations génétiques qui rendent cette enzyme sur-actif, ce qui provoque la codéine en morphine être converti rapidement et plus complètement que dans les autres. Ces métaboliseurs ultra-rapides sont plus susceptibles d'avoir des quantités supérieures à la normale de la morphine dans le sang après la prise de codéine. Des niveaux élevés de morphine peut entraîner des difficultés respiratoires, qui peut être fatale.
De un à sept personnes sur 100 sont métaboliseurs ultra-rapides, mais ils sont plus fréquents chez certains groupes ethniques. Vingt-neuf pour cent des populations d'Afrique du Nord et éthiopiens sont métaboliseurs ultra-rapides, et environ 6 pour cent des Afro-américains, du Caucase et populations grecques sont également touchés.
La seule façon de savoir si quelqu'un est un métabolisme ultra-rapide est de faire un test génétique. Il ya approuvé par la FDA des tests pour vérifier pour le métabolisme ultra-rapide.
Les cas sont survenus chez des enfants présentant des signes d'être métaboliseurs ultra-rapides. Les enfants étaient âgés de 21 mois à 9 ans. Tous les enfants ont reçu des doses de codéine qui étaient dans la gamme de dose typique, ce qui signifie qu'ils n'ont pas eu des quantités supplémentaires de ce médicament.
Dans ces cas, les signes d'une surdose de morphine développés dans un ou deux jours après que les enfants ont commencé à prendre la codéine.
Des signes de troubleLes nouvelles modifications apportées à l'étiquette codéine avertir les professionnels de santé sur l'utilisation de la codéine après une amygdalectomie et / ou adenoidectormy. La FDA affirme que la codéine ne doit être prescrit à des enfants avec d'autres types de douleur si les avantages devraient l'emporter sur les risques.
L'agence avertit que s'ils sont prescrits pour traiter la douleur, la codéine ne doit pas être administré selon un calendrier, mais seulement lorsque l'enfant a besoin de soulagement de la douleur. Les enfants ne devraient jamais recevoir plus de six doses par jour.
Les parents et les soignants doivent surveiller les enfants recevant de la codéine pour la douleur de près les signes de surdose de morphine. Il ya un certain nombre de symptômes à surveiller, dit Bob Rappaport, MD, directeur de la Division des produits d'anesthésie, d'analgésie et des toxicomanies (DAAAP) dans le Centre de la FDA pour l'évaluation des médicaments et de la recherche. Si votre enfant présente ces symptômes, cesser de donner de la codéine et de consulter immédiatement un médecin en prenant votre enfant à la salle d'urgence ou appeler le 911:
- Somnolence inhabituelle, comme étant difficile à réveiller
- Désorientation ou de confusion
- Respiration bruyante ou travaillé, comme respirer superficiellement avec un "soupir" façon de respirer ou respirations profondes séparées par de longues pauses anormalement
- Bleu sur les lèvres ou autour de la bouche
"La chose la plus importante est que les soignants doivent indiquer à l'opérateur 911 ou personnel de service d'urgence que leur enfant a pris de la codéine et a des problèmes respiratoires», dit Rappaport.
Parlez-en à votre professionnel des soins de santé de l'enfant, si vous avez des questions ou des préoccupations concernant la codéine. Si les professionnels de la santé à décider que le bénéfice de la prescription des produits qui contiennent de la codéine à des patients pédiatriques emporte sur le risque, la FDA indiquant que la dose minimale efficace sera prescrite pour la plus courte période de temps.
La réponse aux analgésiques peut être affectée par différents facteurs. De nombreux analgésiques sont métabolisés par l'intermédiaire des isoenzymes du cytochrome P450 dont certains subissent un polymorphisme génétique. Les conséquences cliniques vont de la toxicité médicamenteuse à l'absence d'efficacité selon l'analgésique et le polymorphisme considérés. La pharmacogénétique pourrait permettre à l'avenir d'anticiper, en détectant des polymorphismes génétiques par le génotypage et/ou le phénotypage, mais aussi d'optimaliser et d'individualiser l'approche analgésique médicamenteuse. Elle devrait ainsi permettre d'améliorer la sécurité et l'efficacité des traitements médicamenteux.
Bonus:
(Traduction d'anglais par Google Translator)
 2000-2010 La codéine post-chirurgie met les enfants à risque Les enfants ont souvent prescrits de la codéine pour soulager la douleur après la chirurgie pour enlever leurs amygdales ou des végétations adénoïdes pour traiter l'amygdalite chronique ou d'apnée du sommeil, une condition dans laquelle les problèmes respiratoires font qu'il est difficile pour eux de dormir à poings fermés.
2000-2010 La codéine post-chirurgie met les enfants à risque Les enfants ont souvent prescrits de la codéine pour soulager la douleur après la chirurgie pour enlever leurs amygdales ou des végétations adénoïdes pour traiter l'amygdalite chronique ou d'apnée du sommeil, une condition dans laquelle les problèmes respiratoires font qu'il est difficile pour eux de dormir à poings fermés.Cependant, certains enfants sont morts après avoir reçu la codéine en quantités qui sont dans la fourchette des posologies recommandées.
La Food and Drug Administration (FDA) prend des mesures pour mettre en garde contre l'utilisation de la codéine pour soulager la douleur des enfants après la chirurgie pour enlever leurs amygdales ou des végétations adénoïdes.
En Août 2012, la FDA a averti le public que ce danger existe pour les enfants qui sont des «métaboliseurs ultra-rapides" de la codéine, ce qui signifie que leur foie transforme la codéine en morphine en plus élevée que la normale.
Depuis lors, la FDA a procédé à un examen complet de la sécurité de la codéine chez les enfants. A la recherche de la FDA des événements système de rapports défavorables (SADR) base de données de 1969 à 1 mai 2012 a identifié 10 morts et trois surdoses liés à la codéine. Beaucoup de ces enfants ont été remis d'une intervention chirurgicale pour enlever leurs amygdales ou des végétations adénoïdes.
Fort d'alerte sera une nouvelle boîte d'alerte de la FDA être ajoutées sur l'étiquette des médicaments contenant de la codéine des produits sur le risque de la codéine pour soulager la douleur chez les enfants après une amygdalectomie et / ou adénoïdectomie. (L'étiquette d'un médicament est la documentation accompagnant un médicament d'ordonnance.)
La FDA recommande vivement contre l'utilisation de la codéine pour soulager la douleur chez les enfants après une amygdalectomie et / ou adénoïdectomie. L'agence demande aux professionnels de la santé d'utiliser un autre analgésique.
En outre, les parents et les soignants doivent être conscients des risques d'un traitement codéine après une amygdalectomie ou une adénoïdectomie et doit demander un médicament contre la douleur différente si leur enfant est prescrit codéine dans ce cadre
Le problème
La codéine est un analgésique opioïde analgésique narcotique des médicaments-une-utilisé pour traiter les douleurs légères à modérées. Il est également utilisé afin de réduire la toux, le plus souvent en combinaison avec d'autres médicaments. Codéine est disponible sur ordonnance, seul ou en combinaison avec l'acétaminophène ou de l'aspirine, et dans certains médicaments contre le rhume et la toux.
La codéine est convertie en morphine dans le foie par une enzyme. Certaines personnes ont des variations génétiques qui rendent cette enzyme sur-actif, ce qui provoque la codéine en morphine être converti rapidement et plus complètement que dans les autres. Ces métaboliseurs ultra-rapides sont plus susceptibles d'avoir des quantités supérieures à la normale de la morphine dans le sang après la prise de codéine. Des niveaux élevés de morphine peut entraîner des difficultés respiratoires, qui peut être fatale.
De un à sept personnes sur 100 sont métaboliseurs ultra-rapides, mais ils sont plus fréquents chez certains groupes ethniques. Vingt-neuf pour cent des populations d'Afrique du Nord et éthiopiens sont métaboliseurs ultra-rapides, et environ 6 pour cent des Afro-américains, du Caucase et populations grecques sont également touchés.
La seule façon de savoir si quelqu'un est un métabolisme ultra-rapide est de faire un test génétique. Il ya approuvé par la FDA des tests pour vérifier pour le métabolisme ultra-rapide.
Les cas sont survenus chez des enfants présentant des signes d'être métaboliseurs ultra-rapides. Les enfants étaient âgés de 21 mois à 9 ans. Tous les enfants ont reçu des doses de codéine qui étaient dans la gamme de dose typique, ce qui signifie qu'ils n'ont pas eu des quantités supplémentaires de ce médicament.
Dans ces cas, les signes d'une surdose de morphine développés dans un ou deux jours après que les enfants ont commencé à prendre la codéine.
Des signes de troubleLes nouvelles modifications apportées à l'étiquette codéine avertir les professionnels de santé sur l'utilisation de la codéine après une amygdalectomie et / ou adenoidectormy. La FDA affirme que la codéine ne doit être prescrit à des enfants avec d'autres types de douleur si les avantages devraient l'emporter sur les risques.
L'agence avertit que s'ils sont prescrits pour traiter la douleur, la codéine ne doit pas être administré selon un calendrier, mais seulement lorsque l'enfant a besoin de soulagement de la douleur. Les enfants ne devraient jamais recevoir plus de six doses par jour.
Les parents et les soignants doivent surveiller les enfants recevant de la codéine pour la douleur de près les signes de surdose de morphine. Il ya un certain nombre de symptômes à surveiller, dit Bob Rappaport, MD, directeur de la Division des produits d'anesthésie, d'analgésie et des toxicomanies (DAAAP) dans le Centre de la FDA pour l'évaluation des médicaments et de la recherche. Si votre enfant présente ces symptômes, cesser de donner de la codéine et de consulter immédiatement un médecin en prenant votre enfant à la salle d'urgence ou appeler le 911:
- Somnolence inhabituelle, comme étant difficile à réveiller
- Désorientation ou de confusion
- Respiration bruyante ou travaillé, comme respirer superficiellement avec un "soupir" façon de respirer ou respirations profondes séparées par de longues pauses anormalement
- Bleu sur les lèvres ou autour de la bouche
"La chose la plus importante est que les soignants doivent indiquer à l'opérateur 911 ou personnel de service d'urgence que leur enfant a pris de la codéine et a des problèmes respiratoires», dit Rappaport.
Parlez-en à votre professionnel des soins de santé de l'enfant, si vous avez des questions ou des préoccupations concernant la codéine. Si les professionnels de la santé à décider que le bénéfice de la prescription des produits qui contiennent de la codéine à des patients pédiatriques emporte sur le risque, la FDA indiquant que la dose minimale efficace sera prescrite pour la plus courte période de temps.
Aucun commentaire:
Enregistrer un commentaire