
Généralités
Le métabolisme est l'ensemble des opérations biochimiques qui s'effectuent dans l'organisme. Ces opérations sont réglées par des enzymes. La synthèse de ces enzymes est sous la dépendance des différents gènes portés par les chromosomes.
Lorsqu'à la suite d'une mutation ou d'une transmission héréditaire, il existe une erreur génique, la synthèse de l'enzyme concerné ne se fait plus.
Lorsqu'un enzyme est déficient, le métabolite qui ne peut plus être transformé s'accumule dans le sang et est éliminé en excès dans les urines. Par ailleurs, le produit qui résulte de l'activité normale de l'enzyme déficient n'est pas synthétisé.
La plupart des erreurs innées du métabolisme ont une transmission héréditaire de type autosomique récessif .
Ces affections ne s'expriment qu'après la naissance lorsque l'organisme maternel ne permet plus les processus biochimiques normaux. Dès qu'un aliment ou un médicament donne l'occasion au blocage enzymatique de s'exprimer, la maladie peut s'extérioriser.
Le dépistage précoce de l'affection permettrait d'éviter la rencontre avec le produit qui ne peut être métabolisé, en instituant un régime exclusif. Il permettrait un conseil génétique efficace.
En pratique, ces maladies métaboliques sont recherchées dans trois circonstances différentes. Il peut s'agir du dépistage systématique dans le cadre d'une politique de santé : c'est pour l'instant la pratique obligatoire du test de Guthrie. Il peut s'agir d'une recherche orientée dans une famille à haut risque, lorsqu'un enfant de cette famille est déjà atteint d'une maladie héréditaire. Enfin, l'erreur innée du métabolisme peut être évoquée chez un nouveau-né à l'occasion de troubles cliniques.
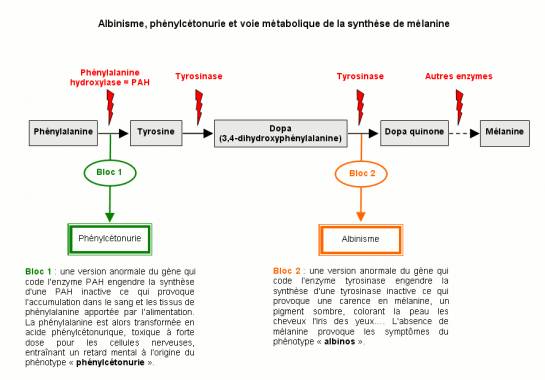
Qu'est-ce que c'est ?
La phénylcétonurie (idiotie phénylpyruvique) est une affection héréditaire transmise sur le mode autosomique récessif. La fréquence de porteurs hétérozygotes dans la population est de 1/50. Un couple sur 2500 présente donc le gène. La fréquence de cette encéphalopathie évolutive dans la population est de 1/16 500.
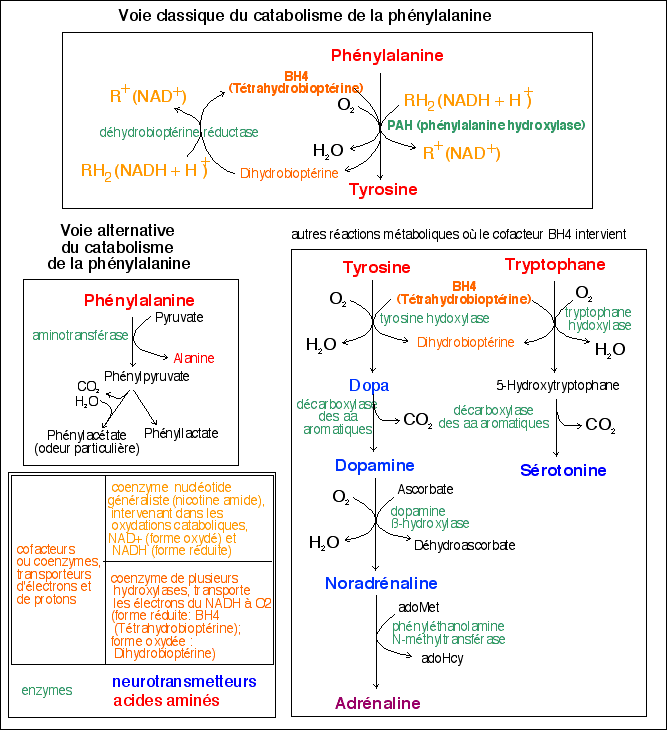
Cette affection est due au déficit en un enzyme hépatique : la phénylalanine-hydroxylase qui permet la transformation de la phénylalanine en tyrosine. Dans la PCU typique, la phénylalanine dans le sang dépasse 25 mg/100 ml.
 Il existe des PCU atypiques où le déficit enzymatique est moins sévère (phénylalanine sanguine entre 12 et 25 mg/100 ml). Ces formes nécessitent quand même un régime restrictif contrôlé. Lorsque le déficit enzymatique est encore moindre (phénylalanine sanguine entre 2 et 12 mg/100 ml), le régime n'est pas nécessaire mais les familles doivent en être informées notamment s'il s'agit d'une fille car la grossesse future devra être surveillée.
Il existe des PCU atypiques où le déficit enzymatique est moins sévère (phénylalanine sanguine entre 12 et 25 mg/100 ml). Ces formes nécessitent quand même un régime restrictif contrôlé. Lorsque le déficit enzymatique est encore moindre (phénylalanine sanguine entre 2 et 12 mg/100 ml), le régime n'est pas nécessaire mais les familles doivent en être informées notamment s'il s'agit d'une fille car la grossesse future devra être surveillée.
Dans 2% des cas, le déficit enzymatique ne concerne pas l'enzyme mais son cofacteur, la tétrahydrobioptérine. Le diagnostic est important car cette forme nécessite un traitement particulier (Dopa etc).
Le test de Guthrie
Quelques gouttes de sang sont prélevées au talon ou au doigt et imbibent une carte en papier buvard qui est envoyée par la poste dans un laboratoire central.
Le laboratoire détruit l'hémoglobine à l'autoclave puis découpe les petites rondelles de papier-buvard imbibées du sang de l'enfant. Ces rondelles sont placées dans une boîte de gélose.
Il s'agit d'un test bactériologique.
Il se trouve en effet que le développement d'une bactérie, le "bacille subtilis" est inhibé sur gélose et que la phénylalanine lève cette inhibition.
Les signes de la maladie
Spontanément très grave, cette maladie est curable par un régime qui doit débuter avant le 3° mois de vie. Seul un diagnostic précoce et un traitement immédiat évitent à ces enfants des troubles neurologiques graves : retard mental, troubles du comportement, psychoses, spasmes en flexion, épilepsie etc.
LE BUT DU REGIMEC’est de contrôler et limiter l’apport en phénylalanine : en apporter juste un peu pour les besoins de croissance du phénylcétonurique, mais sans excès afin de ne pas intoxiquer le cerveau. Ce régime doit par ailleurs couvrir les autres besoins de l’enfant ou adulte en calories, vitamines etc. Il est propre à chaque phénylcétonurique, selon sa tolérance et ses besoins.
LE REGIME
Le régime est mis en place dès le dépistage de la phénylcétonurie. Il est établi par l’équipe médicale qui suit le patient et doit être suivi, tous les jours, à chaque repas sans exception... à vie, selon l’état actuel des connaissances médicales. Pour le moment, il est fortement recommandé de ne jamais complètement abandonner le régime une fois l’âge adulte atteint.
Le régime est constitué de trois types d’aliments pauvres en phénylalanine :
v Les aliments naturels, répartis en trois groupes :
o Les aliments interdits car contenant trop de phénylalanine (viande, poisson, œufs, charcuterie, lait, fromage, laitages…) ainsi que certains aliments à base de céréales (pain, gâteaux, semoule, pâtes…). L’aspartam (faux-sucre) est également interdit car c’est un précurseur de la phénylalanine.
o Les aliments autorisés en quantités pesées en fonction de la tolérance (légumes et fruits).
o Les aliments autorisés à volonté, selon les besoins de l’enfant ou de l’adulte (sucre, miel, confiture, jus de fruit, pâtes de fruits, huile, beurre…).
LE REGIME
Le régime est mis en place dès le dépistage de la phénylcétonurie. Il est établi par l’équipe médicale qui suit le patient et doit être suivi, tous les jours, à chaque repas sans exception... à vie, selon l’état actuel des connaissances médicales. Pour le moment, il est fortement recommandé de ne jamais complètement abandonner le régime une fois l’âge adulte atteint.
Le régime est constitué de trois types d’aliments pauvres en phénylalanine :
v Les aliments naturels, répartis en trois groupes :
o Les aliments interdits car contenant trop de phénylalanine (viande, poisson, œufs, charcuterie, lait, fromage, laitages…) ainsi que certains aliments à base de céréales (pain, gâteaux, semoule, pâtes…). L’aspartam (faux-sucre) est également interdit car c’est un précurseur de la phénylalanine.
o Les aliments autorisés en quantités pesées en fonction de la tolérance (légumes et fruits).
o Les aliments autorisés à volonté, selon les besoins de l’enfant ou de l’adulte (sucre, miel, confiture, jus de fruit, pâtes de fruits, huile, beurre…).

Il y a un certain nombre d’aliments naturels que je ne peux
pas manger, ce sont tous les aliments riches en protéines.
Le complément alimentaire d’acides aminés m’apporte
tout ce dont mon organisme a besoin pour se développer normalement.
Les produits diététiques hypoprotidiques me permettent
de diversifier mes repas et de me faire plaisir.
v Des mélanges d’acides aminés (compléments alimentaires spéciaux, sans phénylalanine), à prendre tous les jours pour compenser le manque de protéines indispensables à la croissance du phénylcétonurique.
v Les produits diététiques hypoprotidiques, appauvris industriellement en phénylalanine. Ce sont des substituts d’aliments naturels qui permettent aux phénylcétonuriques d’avoir une alimentation diversifiée et calorique (pain, pâtes, riz, semoule, soupe, farine, substitut de lait, substitut d’œuf…). Ils ne se trouvent pas dans le commerce et sont commandés par la diététicienne à l’AGEPS (Agence Générale des Equipements et Produits de Santé) qui livre ensuite les produits aux PCU.
Un suivi diététique et médical du phénylcétonurique, avec des examens de contrôles réguliers (cliniques et biologiques), permettent de s’assurer que le régime est bien équilibré et sans carence.
LES DEFIS

Le défi pour les familles au quotidien...
La prise en charge diagnostique et thérapeutique de l’enfant PCU est bien codifiée de nos jours et est fortement individualisée. Le défi médical réside actuellement dans le suivi et la prise en charge des femmes phénylcétonuriques à l’âge adulte : afin de prévenir le risque de handicap chez leurs futurs enfants, elles doivent en effet reprendre un régime strict avant toute grossesse.
Pour les phénylcétonuriques les moins sévèrement atteints, un nouveau type de traitement est apparu ces dernières années. Ce médicament (BH4) permet chez certains patients de retrouver tout ou partie de l'activité du métabolisme afin qu'il régule de lui-même les taux de phénylalanine. Le régime est alors moins contraignant.
BREF HISTORIQUE
. 1934 : le médecin norvégien Yvar Asbjorn Folling est le premier à décrire la phénylcétonurie (PCU).
. 1935 : le généticien anglais nomme la maladie de Folling la "phénylcétonurie"
. 1954 : le professeur allemand Horst Bickel établit le régime pauvre en phénylalanine (une fillette de 3 ans dont le diagnostic de phénylcétonurie avait été fait tard a présenté un comportement nettement amélioré après l'introduction du régime).
. 1961-1963 : le docteur américain Robert Guthrie met au point le dépistage de la PCU par le fameux 'test de Guthrie'.
. 1967 : en France, début de la systématisation du dépistage néonatal de la phénylcétonurie dans quelques départementas (initiative privée de la Société des Eaux d'Evian)
. 1972 : création de l'AFDPHE (Association Française pour le Dépistage des Handicaps de l'Enfant) qui permet d'étendre le dépistage néonatal systématique de la phénylcétonurie à l'ensemble des nouveau-nés des départements français (1978) et Outre-Mer (1979). Le dépistage est pris en charge par la Caisse Nationale de l'Assurance Maladie dès 1972
. 2010 : en France, le dépistage néonatal de la phénylcétonurie, jusqu'alors organisé de manière systématique par l'AFDPHE, devient obligatoire.
. Aujourd'hui : les programme de dépistage de la phénylcétonurie sont une réussite et se mondialisent. les pays européens ont établi des programmes de dépistage similaires et dans les pays en voie de développement, des expériences voient le jour. Il est important de prendre conscience de la nécessité d'un régime alimentaire strict.
Traitement
Le régime pauvre en phénylalanine est prescrit pour environ 8 ans afin de garder un taux de phénylalanine sanguin entre 2 et 5 mg/100 ml. Le régime est ensuite progressivement élargi car après 8 ans, il n'y a plus de risque neurologique même si les anomalies biologiques réapparaissent. Il convient néanmoins de contrôler les apports protéiques pour maintenir la phénylalaninémie au dessous de 16 mg/100 ml.
Lorsqu'il s'agit d'une fille, le régime est repris plus tard en cas de grossesse pour éviter l'embryo-foetopathie phénylalanique qui se traduit chez le nouveau-né par des malformations osseuses, cardiaques et oculaires congénitales, une microcéphalie, une hypotrophie et une arriération mentale.
Kuvan® – Nouveau médicament pour le traitement de la phénylcétonurie
Kuvan ® (saproptérine) est un nouveau médicament indiqué dans le traitement de la phénylcétonurie, une maladie
Kuvan® – Nouveau médicament pour le traitement de la phénylcétonurie
Kuvan ® (saproptérine) est un nouveau médicament indiqué dans le traitement de la phénylcétonurie, une maladie
métabolique rare. L'enzyme qui métabolise la phénylalanine alimentaire dans l'organisme est dysfonctionnelle chez les personnes atteintes de phénylcétonurie. Conséquemment, les concentrations sanguines de phénylalanine sont anormalement élevées, ce qui est toxique pour le cerveau. Sans traitement, la phénylcétonurie peut entraîner des complications neurophysiologiques irréversibles, dont un retard mental, des crises d'épilepsie, un déficit intellectuel,des problèmes comportementaux et sociaux
1,2.
Au Canada, la phénylcétonurie touche 1 nouveau-né sur 12 000 à 15 000. Le dépistage universel de la
phénylcétonurie effectué à la naissance depuis les années 60 permet un dépistage et un traitement précoces. La
phénylcétonurie est maîtrisée par un régime alimentaire strict réduit en protéines alimentaires et un supplément de protéines sans phénylalanine. Kuvan
® est le premier agent pharmacologique approuvé pour le traitement de la
phénylcétonurie. Kuvan
® n'est pas un substitut. Il est plutôt utilisé en association avec un régime sans phénylalanine afin de réduire davantage les concentrations sanguines de phénylalanine. Kuvan® active l'enzyme dysfonctionnelle et rétablit partiellement le métabolisme de la phénylalanine, ce qui a pour effet d'abaisser les concentrations sanguines de phénylalanine1,2.
La dose initiale recommandée est de 10 mg/kg/jour. Si la concentration sanguine initiale de phénylalanine n'a pas diminué après 1 mois, la dose doit être augmentée à 20 mg/kg/jour. Selon les résultats d'études cliniques, de 20 à 56 % des patients répondent au traitement par Kuvan
®. Chez ces patients, la dose quotidienne recommandée varie de 5 à 20 mg/kg/jour. Le coût annuel d'un traitement par Kuvan® varie de 12 045 $ à 156 585 $, en fonction de la dose quotidienne recommandée et du poids des patients allant de 10 à 70 kilogrammes. Le fabricant de Kuvan® offre un programme de mise en route du traitement en fournissant aux patients un premier approvisionnement de 30 jours sans frais, ce qui a pour but de vérifier si les patients qui reçoivent Kuvan® répondent au traitement1.
Parce que son coût risque d'être élevé, Kuvan ® sera exclu des listes de médicaments ouvertes et remboursé en vertu d'une autorisation spéciale si les participants à un régime SécurIndemnité ont souscrit la liste gérée, les programmes de médicaments spécialisés ou le programme de protection en excédent de pertes de SécurIndemnité. L'autorisation spéciale permet à SécurIndemnité de s'assurer de ce qui suit :
1) le médicament est utilisé pour l'indication approuvée,
2) le participant a fait un essai de 30 jours,
3) le participant suit un régime sans phénylalanine,
et 4) la coordination des prestations des programmes provinciaux mis sur pied pour les maladies rares. Si vous désirez que Kuvan
® soit couvert par votre régime collectif, veuillez communiquer avec votre représentant du Service à la clientèle de SécurIndemnité.
Si vous désirez des renseignements supplémentaires au sujet de Kuvan®, veuillez communiquer avec Geneviève Coutu,
pharmacienne-clinicienne, Services cliniques, au 905-949-3031 ou au 1-888-479-7587, poste 3031.
Recommandation :
Exclure/Autorisation spéciale
SécurIndemnité se réserve le droit de modifier en partie ou en entier ses lignes directrices sur ses programmes d'Autorisation Spéciale.
Le 30 avril 2010, Santé Canada a émis à l'intention de BioMarin Pharmaceutical Inc. un avis de conformité du produit pharmaceutique Kuvan.
Kuvan contient l'ingrédient médicinal dichlorhydrate de saproptérine, un activateur enzymatique.
Kuvan est indiqué en association avec un régime à teneur réduite en phénylalanine (PHE) pour réduire la concentration sanguine de PHE chez les patients atteints d'hyperphénylalaninémie (HPA) causée par une phénylcétonurie (PCU) sensible à la tétrahydrobioptérine (BH4).
Kuvan est une formulation synthétique de BH4, un cofacteur de l'enzyme phénylalanine hydroxylase (PAH). La PAH hydroxyle la PHE par une réaction d'oxydation, ce qui produit de la tyrosine. Chez les patients atteints de PCU, l'activité de la PAH est nulle ou insuffisante. Le traitement par BH4 peut augmenter l'activité résiduelle de la PAH, améliorant ainsi le métabolisme oxydatif de la PHE et diminuant la concentration de PHE chez certains patients.
L'autorisation de commercialisation s'appuie sur des données issues d'études de contrôle de la qualité et d'études non cliniques et cliniques. Les études cliniques présentées montrent l'efficacité et l'innocuité cliniques de Kuvan chez les patients atteints de PCU. On a noté une diminution significative de la concentration sanguine de PHE chez les patients recevant Kuvan. De plus, d'après les données sur l'innocuité, le médicament était généralement bien toléré.
Kuvan (100 mg, dichlorhydrate de saproptérine) est offert en comprimés. La dose de départ recommandée de Kuvan est de 10 mg/kg/jour. On évalue la réponse au traitement se basant sur le changement de la concentration sanguine de PHE après l'administration de 10 mg/kg/jour de Kuvan pendant une période pouvant aller jusqu'à 1 mois. La concentration sanguine de PHE devrait être vérifiée après une semaine de traitement par Kuvan et de manière périodique pendant une période pouvant atteindre 1 mois. Si la concentration sanguine de PHE ne descend pas sous la valeur initiale après l'administration de 10 mg/kg/jour, on peut augmenter la dose chaque semaine jusqu'à un maximum de 20 mg/kg/jour, en surveillant de près la concentration sanguine de PHE pendant 1 mois. On considère que les patients dont la concentration sanguine de PHE ne diminue pas après un traitement de 1 mois à 20 mg/kg/jour ne répondent pas au traitement; l'administration de Kuvan devrait être interrompue chez ces patients. Une fois qu'on a établi que le patient répond à Kuvan, la posologie peut être ajustée dans une fourchette de 5 à 20 mg/kg/jour en fonction de la réponse au traitement. Dans les études cliniques, on n'a pas évalué de doses de Kuvan supérieures à 20 mg/kg/jour. Les recommandations concernant la posologie sont énoncées dans la monographie de produit.
Kuvan est contre-indiqué chez les patients présentant une hypersensibilité au produit, à un ingrédient de la formulation ou à un composant du contenant. Kuvan devrait être administré selon les conditions décrites dans la monographie de produit, en tenant compte des risques potentiels associés à l'administration de ce produit pharmaceutique. Les conditions détaillées relatives à l'usage de Kuvan sont décrites dans la monographie de produit.
Kuvan a été évalué dans le cadre de la Politique sur le traitement prioritaire, car il semblait offrir un traitement efficace d'une maladie ou d'une affection grave, potentiellement fatale ou sévèrement débilitante, pour laquelle aucun médicament n'est actuellement commercialisé au Canada.
Conformément à son examen des données portant sur la qualité, l'innocuité et l'efficacité du produit, Santé Canada juge que le rapport entre les avantages et les risques de Kuvan est favorable pour l'indication ci-dessus.
Renseignements généraux
Le dichlorhydrate de saproptérine, l'ingrédient médicinal de Kuvan, est un activateur enzymatique. Chez les patients atteints de phénylcétonurie (PCU), l'activité de l'enzyme phénylalanine hydroxylase (PAH) est nulle ou insuffisante, ce qui entraîne une augmentation de la concentration sanguine de phénylalanine (PHE). Il est établi que le dichlorhydrate de saproptérine stimule l'activité de la PAH endogène et le rétablissement partiel du métabolisme oxydatif de la PHE, d'où une diminution de la concentration sanguine de PHE chez les patients atteints de PCU.Procédé de fabrication et contrôles en cours de fabrication
Le dichlorhydrate de saproptérine est fabriqué par une synthèse en plusieurs étapes. Chaque étape du procédé de fabrication est considérée comme contrôlée à l'intérieur de limites acceptables :- Le promoteur a fourni de l'information sur la qualité et les contrôles pour toutes les matières qui entrent dans la fabrication de la substance médicamenteuse.
- Les spécifications de la substance médicamenteuse sont jugées satisfaisantes. Les valeurs limites quant aux impuretés sont conformes aux exigences de l'International Conference on Harmonisation (ICH).
- Les étapes de traitement ont été évaluées et des intervalles appropriés ont été établis relativement aux paramètres des procédés.
Caractérisation
La structure du dichlorhydrate de saproptérine a été adéquatement élucidée, et les spectres représentatifs ont été fournis. Les propriétés physiques et chimiques ont été décrites et sont jugées satisfaisantes.Les impuretés et produits de dégradation découlant de la fabrication et/ou de l'entreposage ont été signalés et caractérisés. Ces produits étaient conformes aux limites établies par l'ICH et/ou ont été qualifiés au moyen d'analyses de lots; ils sont donc considérés comme acceptables.
Contrôle de la substance médicamenteuse
Les spécifications de la substance médicamenteuse ainsi que les méthodes d'analyse utilisées pour le contrôle de la qualité du dichlorhydrate de saproptérine ont été jugées acceptables.Après examen, on a déterminé que les résultats des analyses de lots sont tous conformes aux spécifications et témoignent d'une qualité constante des lots produits.
L'emballage de la substance médicamenteuse est jugé acceptable.
Stabilité
Les résultats d'études de stabilité fondées sur des essais accélérés, de longue durée et sous contrainte montrent que le dichlorhydrate de saproptérine est un composé stable lorsqu'il est conditionné de la façon prévue pendant la période d'entreposage proposée. Le médicament en vrac est également stable dans les conditions d'entreposage prévues.Absorption
La saproptérine est principalement absorbée par l'intestin grêle, y compris le duodénum. Le délai pour atteindre la concentration plasmatique maximale (Tmax) était de 1 à 3 heures après l'administration orale chez la souris, le rat et le singe. La demi-vie était de 1,2, 1,1 et 1,4 heure chez la souris, le rat et le singe, respectivement. La biodisponibilité était de 7 % à 12 % chez le rat et d'environ 9 % chez le singe.La saproptérine ne s'est pas accumulée chez le rat après l'administration de doses multiples de 100 mg/kg/jour pendant 14 jours. La cinétique de l'absorption et de l'élimination est demeurée relativement inchangée après l'administration de doses orales multiples.
Distribution
Des études ont révélé que la saproptérine était principalement diffusée dans les reins et le foie. On n'a noté aucune élévation significative de la saproptérine dans le placenta des rates gravides et le fœtus, par rapport aux concentrations endogènes observées chez cette espèce. Bien que les concentrations de saproptérine aient augmenté dans les glandes mammaires des rates mères, aucune hausse n'a été observée dans le lait maternel à la dose de 10 mg/kg.Les études sur la liaison aux protéines plasmatiques chez le rat (in vivo) et l'humain (in vitro) n'ont fait ressortir aucune liaison notable de la bioptérine totale aux protéines plasmatiques.
Métabolisme
Des études sur les microsomes hépatiques de rat ont montré que la saproptérine n'activait pas la voie métabolique du cytochrome P450. Ce sont plutôt les enzymes dihydrofolate réductase et dihydroptéridine réductase qui sont responsables du métabolisme et du recyclage de la saproptérine.Selon la littérature scientifique, les voies cataboliques du rat et de l'humain sont semblables. La tétrahydrobioptérine (6R-BH4 [médicament présumé inchangé]), la dihydrobioptérine, la bioptérine, la ptérine et la 6-hydroxylumazine ont été les principaux métabolites urinaires détectés chez le rat après l'administration intraveineuse de 10 mg/kg de saproptérine.
Excrétion
Après l'administration orale de saproptérine radiomarquée à des rats, environ 7 % de la dose radioactive administrée a été éliminée dans l'urine et environ 75 % a été éliminée dans les selles en 72 heures.
Aucun commentaire:
Enregistrer un commentaire