
 En 1992, à l’âge de 42 ans, ce patient avait subi de nombreuses investigations en raison d’épigastralgies, de céphalées, d’une intolérance à certains aliments et de tests hépatiques perturbés.
En 1992, à l’âge de 42 ans, ce patient avait subi de nombreuses investigations en raison d’épigastralgies, de céphalées, d’une intolérance à certains aliments et de tests hépatiques perturbés.
L’échographie abdominale avait révélé une cholestérolose vésiculaire avec un petit polype et l’œsogastroduodénoscopie (OGD) une discrète œsophagite de reflux et une gastrite érosive antrale.
Le patient a alors subi une cholécystectomie et une ponction-biopsie hépatique (PBF) par voie laparoscopique. L’examen anatomopathologique a conclu à une cholestérolose de la vésicule biliaire et à une stéatose hépatique (50% des hépatocytes, avec légère inflammation banale et légère hémosidérose, sans signe d’éthylisme chronique), un diagnostic correspondant à ce que nous appelons actuellement stéatohépatite non alcoolique (NASH).
A suivi une période peu symptomatique, avec pour traitement un régime seul (diminution des produits laitiers, suppression de l’alcool, augmentation des fruits, utilisation d’huile de colza).
Le bilan biologique de février 2002 a permis d’exclure une hépatite A, B ou C, mais montré une bilirubine toujours légèrement élevée et un taux de prothrombine (TP) spontané à 64%.
 En 2007, sont apparus des symptômes mêlant céphalées postprandiales, intolérance alimentaire et, occasionnellement, des épisodes de diarrhées. Des épigastralgies ont motivé une nouvelle OGD qui a révélé une œsophagite de reflux discrète et une gastrite biliaire (de signification clinique controversée selon le gastro-entérologue).
Malgré un traitement d’Ulcogant 4 x/jour, des douleurs abdominales intenses, récidivantes, ont motivé un bilan complémentaire (tableau 1).
En 2007, sont apparus des symptômes mêlant céphalées postprandiales, intolérance alimentaire et, occasionnellement, des épisodes de diarrhées. Des épigastralgies ont motivé une nouvelle OGD qui a révélé une œsophagite de reflux discrète et une gastrite biliaire (de signification clinique controversée selon le gastro-entérologue).
Malgré un traitement d’Ulcogant 4 x/jour, des douleurs abdominales intenses, récidivantes, ont motivé un bilan complémentaire (tableau 1).
 Le CT-scan abdominal, l’échographie, puis l’IRM ont montré plusieurs nodules hépatiques suggérant une stéatose ou des adénomes multiples. Une PBF programmée au début de l’année suivante a dû être repoussée en raison d’un TP spontané à 60% (malgré l’administration de Konakion PO avant l’intervention).
Le CT-scan abdominal, l’échographie, puis l’IRM ont montré plusieurs nodules hépatiques suggérant une stéatose ou des adénomes multiples. Une PBF programmée au début de l’année suivante a dû être repoussée en raison d’un TP spontané à 60% (malgré l’administration de Konakion PO avant l’intervention).
Le patient a alors été adressé à un hématologue qui note une diminution des facteurs VII et X, compatible avec un déficit en vitamine K et retient le diagnostic vraisemblable de malabsorption. Une hémochromatose a été exclue par dosage génétique. Des injections de Konakion IV ont permis d’augmenter le TP et d’effectuer une PBF sous contrôle échographique. Celle-ci a révélé une hépatite chronique avec une activité inflammatoire légère à modérée, un remaniement hépatique fibreux et de larges plages de dysplasie à petites cellules.
 Les plaintes du patient, associant nausées, vomissements, crampes abdominales violentes et parfois diarrhées, étant toujours présentes, un traitement de Créon, Légalon, Hépa-S, spasmolytiques et inhibiteurs de la pompe à protons a été introduit, sans réelle efficacité.
Le patient a alors été adressé à un autre gastro-entérologue qui a retenu, parmi les diagnostics possibles pour les douleurs abdominales, une possible migraine atypique. Le patient a ensuite été envoyé vers un neurologue qui a conclu à des céphalées d’allure mixte, migraineuses et tensionnelles, et proposé un traitement de métamizole puis, en cas d’inefficacité, d’élétriptan. Ces deux traitements n’ont pas modifié les symptômes.
Les plaintes du patient, associant nausées, vomissements, crampes abdominales violentes et parfois diarrhées, étant toujours présentes, un traitement de Créon, Légalon, Hépa-S, spasmolytiques et inhibiteurs de la pompe à protons a été introduit, sans réelle efficacité.
Le patient a alors été adressé à un autre gastro-entérologue qui a retenu, parmi les diagnostics possibles pour les douleurs abdominales, une possible migraine atypique. Le patient a ensuite été envoyé vers un neurologue qui a conclu à des céphalées d’allure mixte, migraineuses et tensionnelles, et proposé un traitement de métamizole puis, en cas d’inefficacité, d’élétriptan. Ces deux traitements n’ont pas modifié les symptômes.
Finalement, en 2008, sur la base d’une céruloplasmine non mesurable, d’une cuprurie de 24 heures augmentée et d’une analyse génétique (trois mutations du gène ATP7B), le diagnostic de maladie de Wilson a été posé à l’Inselspital, à Berne.
L’évolution a été favorable sous traitement oral de pénicillamine Da associée à la vitamine B6b, ce qui a permis de stabiliser les valeurs biologiques.
Enfin, au début 2012, le patient qui prenait régulièrement son traitement de pénicillamine D a présenté une récidive de douleurs abdominales intenses, avec nausées et vomissements. Le bilan complémentaire (échographie et CT-scan) a posé le diagnostic de carcinome hépatocellulaire multifocal (évolution extrêmement rare dans le cadre d’une maladie de Wilson) et motivé une résection hépatique atypique en février de la même année.
Après une période postopératoire difficile, marquée par une importante perte pondérale, l’état général s’est amélioré. Le traitement de pénicillamine D a été poursuivi.
La céruloplasmine est typiquement abaissée, mais elle peut être faussement normale en cas d’inflammation ou d’hyperœstrogénisme (grossesse ou œstrogénothérapie par exemple). Son dosage ne peut donc, à lui seul, permettre de poser ou d’infirmer le diagnostic de maladie de Wilson.
Idéalement, un diagnostic moléculaire de la mutation ATP7B présente chez le parent index est recommandé. Si ce test n’est pas disponible, il est recommandé de faire un examen clinique à la recherche de signes d’hépatopathie, de troubles neurologiques et/ou d’un anneau de Kayser-Fleischer (lampe à fente par un ophtalmologue). Le bilan biologique inclut la fonction hépatique (TP, albumine, bilirubine totale et directe), les tests hépatiques de cytolyse et de cholestase (ASAT, ALAT, phosphatase alcaline et γGT) et un dosage de la céruloplasmine.
Le traitement initial des patients symptomatiques doit inclure un agent chélateur (pénicillamine D ou trientine). Le traitement des patients asymptomatiques ou ayant une manifestation neurologique seulement peut être effectué soit par un agent chélateur, soit par du zinc. La pénicillamine D (750-1500 mg/jour) doit être prise en 2-3 doses, une heure avant les repas, avec une supplémentation de pyridoxine (25-50 mg/jour). La trientine (900-2700 mg/jour) se prend également en 2-3 doses quotidiennes, une heure avant ou trois heures après les repas. Le zinc (150 mg/jour) se prend 3 x/jour 30 minutes avant les repas.
Le patient a alors subi une cholécystectomie et une ponction-biopsie hépatique (PBF) par voie laparoscopique. L’examen anatomopathologique a conclu à une cholestérolose de la vésicule biliaire et à une stéatose hépatique (50% des hépatocytes, avec légère inflammation banale et légère hémosidérose, sans signe d’éthylisme chronique), un diagnostic correspondant à ce que nous appelons actuellement stéatohépatite non alcoolique (NASH).
A suivi une période peu symptomatique, avec pour traitement un régime seul (diminution des produits laitiers, suppression de l’alcool, augmentation des fruits, utilisation d’huile de colza).
Le bilan biologique de février 2002 a permis d’exclure une hépatite A, B ou C, mais montré une bilirubine toujours légèrement élevée et un taux de prothrombine (TP) spontané à 64%.
En 2007, sont apparus des symptômes mêlant céphalées postprandiales, intolérance alimentaire et, occasionnellement, des épisodes de diarrhées. Des épigastralgies ont motivé une nouvelle OGD qui a révélé une œsophagite de reflux discrète et une gastrite biliaire (de signification clinique controversée selon le gastro-entérologue).
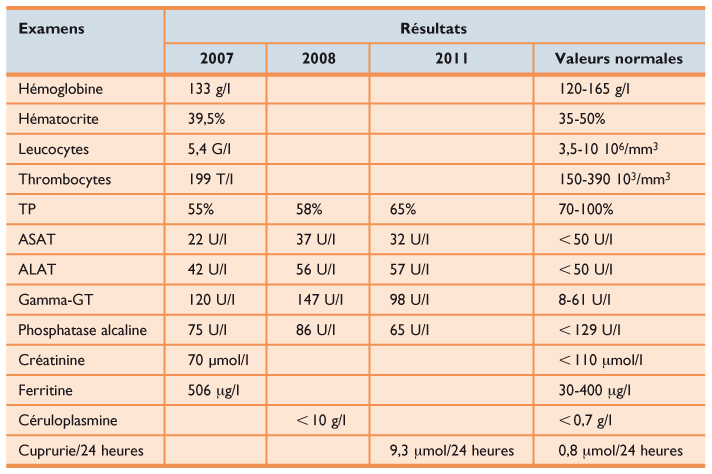
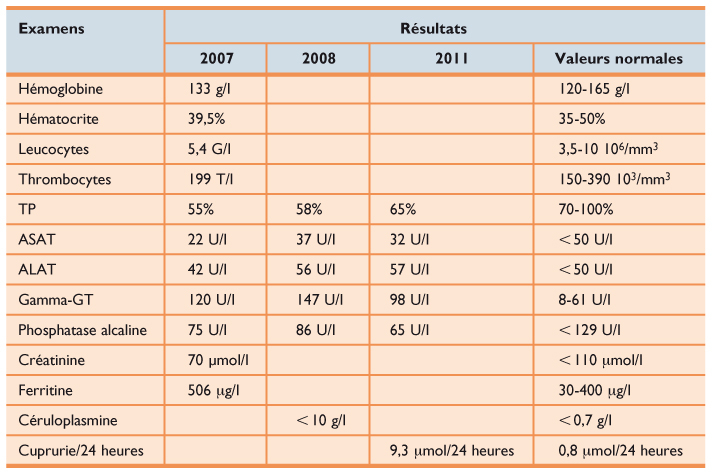
Tableau 1
Bilans biologiques successifs
Examens Résultats 2007 2008 2011 Valeurs normales Hémoglobine 133 g/l 120-165 g/l Hématocrite 39,5% 35-50% Leucocytes 5,4 G/l 3,5-10 106/mm3 Thrombocytes 199 T/l 150-390 103/mm3 TP 55% 58% 65% 70-100% ASAT 22 U/l 37 U/l 32 U/l < 50 U/l ALAT 42 U/l 56 U/l 57 U/l < 50 U/l Gamma-GT 120 U/l 147 U/l 98 U/l 8-61 U/l Phosphatase alcaline 75 U/l 86 U/l 65 U/l < 129 U/l Créatinine 70 μmol/l < 110 μmol/l Ferritine 506 μg/l 30-400 μg/l Céruloplasmine < 10 g/l < 0,7 g/l Cuprurie/24 heures 9,3 μmol/24 heures 0,8 μmol/24 heures
Le patient a alors été adressé à un hématologue qui note une diminution des facteurs VII et X, compatible avec un déficit en vitamine K et retient le diagnostic vraisemblable de malabsorption. Une hémochromatose a été exclue par dosage génétique. Des injections de Konakion IV ont permis d’augmenter le TP et d’effectuer une PBF sous contrôle échographique. Celle-ci a révélé une hépatite chronique avec une activité inflammatoire légère à modérée, un remaniement hépatique fibreux et de larges plages de dysplasie à petites cellules.
Les plaintes du patient, associant nausées, vomissements, crampes abdominales violentes et parfois diarrhées, étant toujours présentes, un traitement de Créon, Légalon, Hépa-S, spasmolytiques et inhibiteurs de la pompe à protons a été introduit, sans réelle efficacité.Finalement, en 2008, sur la base d’une céruloplasmine non mesurable, d’une cuprurie de 24 heures augmentée et d’une analyse génétique (trois mutations du gène ATP7B), le diagnostic de maladie de Wilson a été posé à l’Inselspital, à Berne.
L’évolution a été favorable sous traitement oral de pénicillamine Da associée à la vitamine B6b, ce qui a permis de stabiliser les valeurs biologiques.
Enfin, au début 2012, le patient qui prenait régulièrement son traitement de pénicillamine D a présenté une récidive de douleurs abdominales intenses, avec nausées et vomissements. Le bilan complémentaire (échographie et CT-scan) a posé le diagnostic de carcinome hépatocellulaire multifocal (évolution extrêmement rare dans le cadre d’une maladie de Wilson) et motivé une résection hépatique atypique en février de la même année.
Après une période postopératoire difficile, marquée par une importante perte pondérale, l’état général s’est amélioré. Le traitement de pénicillamine D a été poursuivi.
Questions au spécialiste
Faut-il systématiquement doser la céruloplasmine en cas de stéatohépatite non alcoolique (NASH) ou d’hépatopathie d’origine peu claire ?
Une maladie de Wilson doit être considérée chez toute personne présentant une perturbation des tests hépatiques d’origine indéterminée, avec ou sans troubles neurologiques ou neuropsychiatriques associés. Si cela est surtout recommandé pour les personnes entre 3 et 55 ans, l’âge seul ne doit pas être un critère d’exclusion de la maladie. La maladie doit également être suspectée en cas d’hépatite auto-immune atypique ou répondant mal au traitement, lors de lésions histologiques évoquant une NASH ou une hépatite alcoolique, lors d’une insuffisance hépatique aiguë associée à une hémolyse intravasculaire Coombs négative, avec une élévation modérée des transaminases ou une phosphatase alcaline basse avec un rapport phosphatase alcaline/bilirubine < 2.La céruloplasmine est typiquement abaissée, mais elle peut être faussement normale en cas d’inflammation ou d’hyperœstrogénisme (grossesse ou œstrogénothérapie par exemple). Son dosage ne peut donc, à lui seul, permettre de poser ou d’infirmer le diagnostic de maladie de Wilson.
Quelle attitude adopter envers les frères et sœurs ou les descendants d’une personne atteinte de la maladie de Wilson ? Un dépistage ou des tests génétiques doivent-ils être effectués chez les proches ?
Le dépistage des parents du 1er degré est fortement recommandé. La fratrie d’un patient homozygote a environ 25% de risque de développer une maladie clinique. Ce risque n’est que de 0,5% chez les enfants mais, en raison de la sévérité de la maladie, un diagnostic précoce est justifié.Idéalement, un diagnostic moléculaire de la mutation ATP7B présente chez le parent index est recommandé. Si ce test n’est pas disponible, il est recommandé de faire un examen clinique à la recherche de signes d’hépatopathie, de troubles neurologiques et/ou d’un anneau de Kayser-Fleischer (lampe à fente par un ophtalmologue). Le bilan biologique inclut la fonction hépatique (TP, albumine, bilirubine totale et directe), les tests hépatiques de cytolyse et de cholestase (ASAT, ALAT, phosphatase alcaline et γGT) et un dosage de la céruloplasmine.
Quel traitement/régime proposer une fois le diagnostic confirmé ?
Une fois le diagnostic établi, un régime pauvre en cuivre et un traitement médicamenteux doivent être introduits et poursuivis à vie en l’absence de transplantation hépatique. Les aliments riches en cuivre sont les fruits de mer, les noix, le chocolat, les champignons et les abats. L’eau circulant dans des canalisations en cuivre ne peut être consommée et il convient d’éviter de cuisiner dans des récipients en cuivre.Le traitement initial des patients symptomatiques doit inclure un agent chélateur (pénicillamine D ou trientine). Le traitement des patients asymptomatiques ou ayant une manifestation neurologique seulement peut être effectué soit par un agent chélateur, soit par du zinc. La pénicillamine D (750-1500 mg/jour) doit être prise en 2-3 doses, une heure avant les repas, avec une supplémentation de pyridoxine (25-50 mg/jour). La trientine (900-2700 mg/jour) se prend également en 2-3 doses quotidiennes, une heure avant ou trois heures après les repas. Le zinc (150 mg/jour) se prend 3 x/jour 30 minutes avant les repas.
Quels contrôles effectuer et à quelle fréquence une fois le diagnostic posé et le traitement instauré ?
En début de traitement, le type et la fréquence des contrôles dépendent de la substance utilisée. Une fois la maladie stabilisée, le suivi de routine est biannuel. Il doit comprendre un taux de cuivre sérique total, le cuivre sérique non lié à la céruloplasmine, la céruloplasmine, les tests hépatiques de cytolyse et de cholestase, le TP, l’albumine, la bilirubine totale et directe, une formule sanguine avec répartition, une cuprémie de 24 heures sous traitement et éventuellement aussi 48 heures après l’arrêt du traitement (pour la pénicillamine D et la trientine). En cas de cirrhose ou de fibrose avancée, un dépistage précoce du carcinome hépatocellulaire est également nécessaire au moyen d’un ultra-son hépatique ± un dosage de l’α-fœtoprotéine.
Les écarts de régime occasionnels peuvent-ils poser problème (par exemple : envie de manger comme les autres au restaurant) ?
Les aliments riches en cuivre doivent être strictement évités durant la première année de traitement. Par la suite, et seulement si le patient est stable, une consommation très occasionnelle et en petite quantité de ces aliments peut être tolérée.
Le traitement et le pronostic du carcinome hépatocellulaire survenant dans le cadre d’une maladie de Wilson sont-ils similaires à ceux des formes «classiques» ?
Si la fréquence d’apparition d’un carcinome hépatocellulaire lors d’une cirrhose ou d’une fibrose avancée sur maladie de Wilson est plus faible que dans d’autres hépatopathies chroniques, le traitement et le pronostic sont les mêmes. Les lésions de petite taille, en faible nombre, sans invasion vasculaire ou métastases extra-hépatiques ayant un bien meilleur pronostic, un dépistage précoce de carcinome hépatocellulaire est recommandé chez ces patients. Un bénéfice sur la survie des patients a en effet été démontré, quelle que soit la maladie hépatique sous-jacente.
Aucun commentaire:
Enregistrer un commentaire